MINI-REVIEW
Fabry disease: Spotting the zebra amongst horses

By Dr. Anoushka Krishnan
Nephrologist
GlomCon editors who contributed significantly to the development of this article include Paolo Nikolai So, MD, and Nasim Wiegley, MD.
Introduction
Fabry disease is a rare progressive multisystemic lysosomal storage disease. This X-linked genetic disorder often involves the kidneys in early life, with high risk of progression to end-stage kidney disease (ESKD) in almost all men and some women. It is often managed by multi-disciplinary teams involving cardiologists, nephrologists, neurologists, pain specialists and psychiatrists. Here, we aim to review some key points on disease manifestation, genetics, diagnosis and treatment.
Pathophysiology
Fabry disease is an X-linked lysosomal storage disorder that affects multiple organs. Typically, the genetic defect manifests as a reduced or deficient activity of alpha-galactosidase A (alpha-GAL), an enzyme encoded by the GLA gene on chromosome Xq22, responsible for breaking down glycosphingolipids (fatty acids) within cell lysosomes. Due to a lack of this enzyme, there is gradual accumulation of these glycosphingolipids, particularly, a molecule called globo-triaocylceramide (GB3 or GL3) and its deactivated derivative globo-triaocylsphingosine (lyso-GL3 or lyso-GB3). Deposition of these substances in various organs results in varied disease manifestations.
Prevalence and genetics
Fabry disease has a prevalence of approximately 1 in 117,000 live births for males. It is estimated to affect ~1 in 40,000 males and ~1 in 20,000 females. Its genetic inheritance is well described with the GLA gene being sequenced and hundreds of mutations identified.
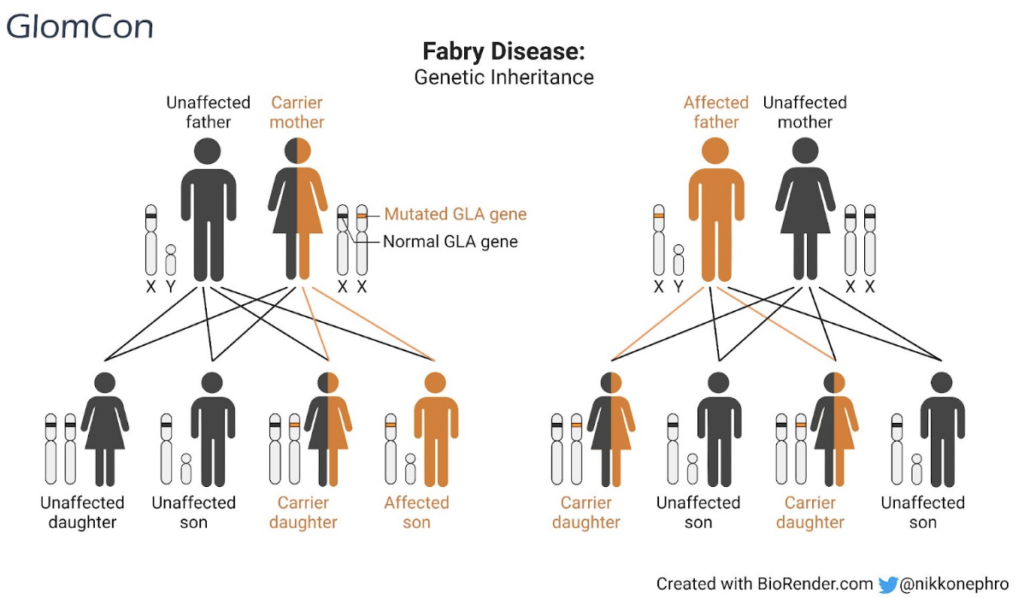
The disease is inherited in an X-linked recessive manner; males can inherit the defective gene from their carrier mothers and can pass on the affected gene to all their daughters who are heterozygotes but not to their sons. Females with the disease are heterozygote for the mutation and can acquire the defective gene from either parent. They have a 50% chance of passing the gene on to their daughters or sons. Generally speaking, males are more severely affected than female heterozygotes (Figure 1).
Figure 1. X-linked recessive genetic inheritance of Fabry disease
Classic Fabry disease is the most severe form of the disease. It is seen in males and has little to no functional alpha-GAL enzyme activity (<1%) with resultant severe multi-organ involvement. Female heterozygotes may have a variable disease course ranging from asymptomatic disease to a more severe phenotype (due to lyonization or random-X inactivation). The ‘late-onset’ phenotype usually presents after the 4th decade, with variable alpha-GAL levels (2-30%) and often with less severe disease.
While over 1000 mutations have been identified, most families have a specific mutation and de novo mutations are rare. Mutations that result in little to no alpha-GAL production result in a severe phenotype while those with some residual alpha-GAL may have milder disease.
Clinical manifestations
As mentioned, clinical manifestations of the disease can be varied and can affect multiple organ systems (Figure 2). Typically, kidney manifestations include proteinuria, micro-hematuria, isosthenuria, hypertension, parapelvic cysts on kidney imaging and ultimately chronic kidney disease. GB3 accumulates preferentially in the glomeruli (especially in podocytes) resulting in excessive podocyte loss and the resultant early clinical manifestation of proteinuria. GB3 can also accumulate in distal tubular cells (resulting in reduced urine concentrating ability), vascular smooth muscle cells and rarely in proximal tubules (Fanconi’s syndrome). Kidney disease can begin during late adolescence, whereas about 50% of males with the classic Fabry phenotype will develop kidney manifestations in their 30s.
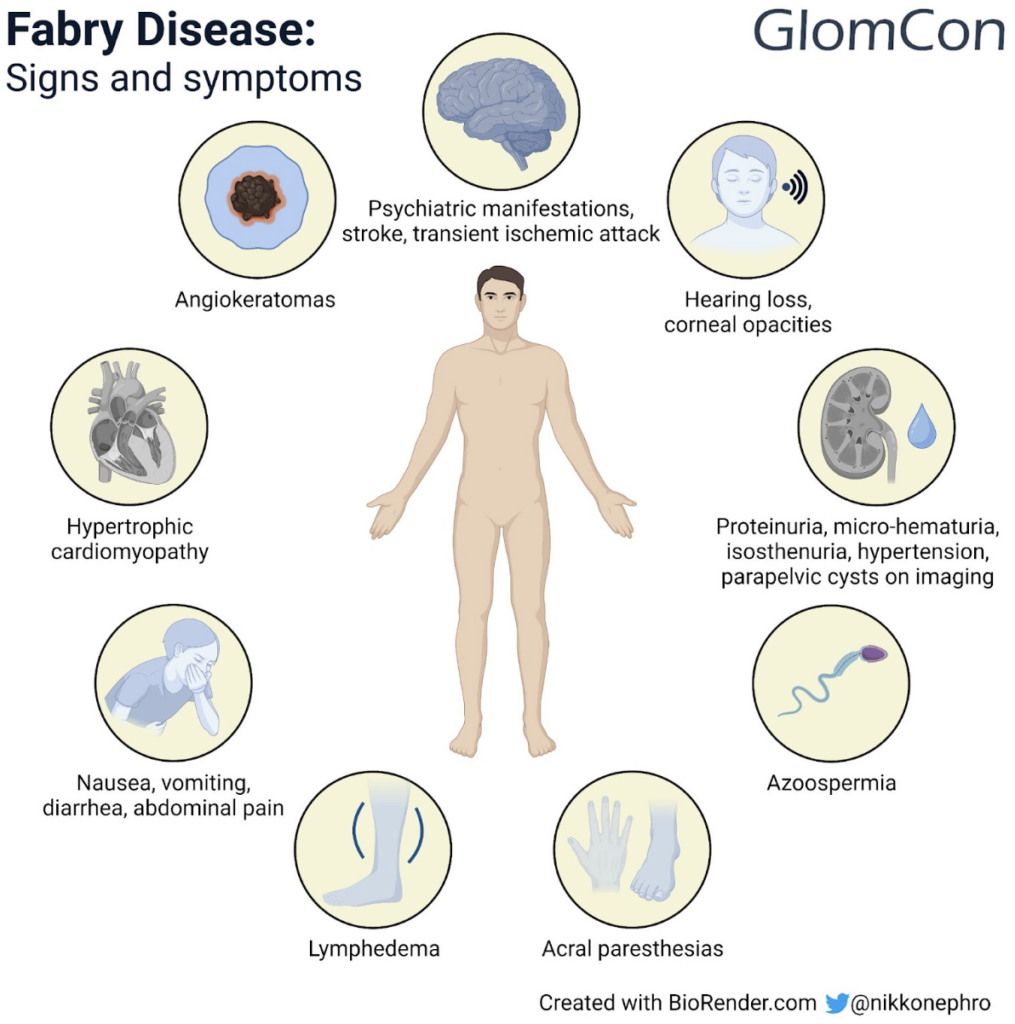
Figure 2. Clinical manifestations of Fabry disease
Pain is a common early and disabling manifestation of the disease. Patients typically report acral paresthesias and can develop pain crises that can be triggered by hot weather, stress or physical illness.
Angiokeratomas are the most common and classic skin manifestation of Fabry disease. These are purplish-red lesions that develop across the umbilicus, groin and upper thighs. Other skin findings include telangiectasias and hypohydrosis.
Cardiac complications are well described in Fabry disease with the most common manifestation being that of hypertrophic cardiomyopathy. Other changes include left ventricular hypertrophy, mitral valve regurgitation, diastolic heart failure, ischemia and cardiac arrhythmias.
The frequency of strokes and transient ischemic attacks is much higher in this population compared to the general population. Gastrointestinal (GI) manifestations can include nausea, vomiting, diarrhea and abdominal pain all of which have significant impacts on quality of life (QoL) in these patients. Additionally, hearing loss is more common in those with Fabry disease (usually sensori-neural in nature) compared to the age-matched general population. Psychiatric manifestations, corneal opacities, lymphedema and azoospermia may also occur.
Diagnosis
Diagnosis is often delayed due to varied and often non-specific symptoms. However, once suspected, the diagnosis can be reliably made by checking for alpha-GAL levels in plasma or peripheral leukocytes (which is low or undetectable in Fabry disease) and by genetic sequencing of the GLA gene (to identify mutations). Detection of elevated levels of lyso-CTH (lyso-ceramide trihexoside) can further confirm the diagnosis and this can be used to monitor response to enzyme replacement therapy or oral chaperone therapy.
A basic blood panel should include troponin and BNP (brain natriuretic peptide) levels apart from blood counts, kidney function and liver function tests. Urine should be evaluated for microscopy and quantification of proteinuria. Electrocardiogram, echocardiogram, cardiac and brain MRI (magnetic resonance imaging), audiometry, pain evaluation and QoL questionnaires are often part of initial work-up and need to be repeated every 1-3 years as clinically indicated.
Kidney biopsy is indicated in cases of declining kidney function or urinary abnormalities. Kidney biopsy is diagnostic with light microscopy demonstrating accumulation of GB3 in podocytes and distal tubular cells with smaller amounts scattered in the mesangium, endothelium, proximal tubule and peritubular capillaries. Other non-specific changes in advanced disease include glomerulosclerosis, interstitial fibrosis and tubular atrophy. Notably, the glomeruli on the biopsy sample may appear white (rather than red) to the naked eye. Electron microscopy classically shows ‘zebra bodies’ most prominently in the podocytes and distal tubules – these represent the accumulated GB3 within lysosomes which appear as lamellated structures.
Management
Treatment is often multi-pronged with multiple specialists involved in providing care. Management focuses on symptom control, Fabry-specific therapy and surveillance of organ systems.
Fabry-specific treatment includes enzyme replacement therapy (ERT) and oral chaperone therapy (Table 1). ERT refers to humanized alpha-GAL and is currently available in two formulations, namely agalsidase alpha (Replagal) and agalsidase beta (Fabrazyme), which can often be administered at home by trained nursing staff. Adverse effects commonly include infusion reactions and rarely the development of neutralizing antibodies to agalsidase; the latter is manifested by loss of ERT efficacy and rising lyso-CTH substrate levels in blood. ERT has been shown to reduce symptom burden (pain, GI symptoms), perhaps stabilize left ventricular mass and clear substrate from kidneys.
Migalastat is an oral chaperone therapy that can be used in certain amenable GLA gene mutations (this can be confirmed via the Galafold amenability website). These patients are able to make some alpha-GAL enzyme that is capable of degrading substrate. However, because of a genetic mutation, the produced enzyme is not effectively delivered to lysosomes. Migalastat acts by binding to the site of the mutated enzyme, stabilizing it and allowing its passage into the lysosomes. The drug then dissociates from the enzyme, thereby allowing alpha-GAL to catabolize the substrate.
Table 1. Comparison of Fabry-specific therapies in terms of dosing, monitoring, and impact on kidney disease
|
Fabry-specific therapy |
Enzyme replacement therapy (ERT) |
Oral chaperone therapy |
|---|---|---|
|
Dosing |
|
Maybe used for amenable mutations Migalastat 150 mg orally on alternate days |
|
Monitoring |
Can use Lyso-CTH levels to monitor response to treatment (decreased levels expected with commencement of therapy) |
Recheck Alpha-GAL levels within 3 months of commencing therapy to demonstrate efficacy (rise in alpha-GAL levels expected) |
|
Impact on kidney disease |
High quality data lacking on progression to end-stage kidney disease |
Not approved for advanced kidney disease (i.e., estimated glomerular filtration rate <30ml/min/1.73m2) |
Gene therapy trials that could potentially be curative are currently being studied across the world (Search of: Fabry Disease – List Results – ClinicalTrials.gov).
All patients should be offered supportive care and symptom management. Renin-angiotensin-aldosterone inhibitors should be used for their kidney protective and anti-proteinuric benefits. Chronic kidney disease, its associated complications and preparation for kidney replacement therapy are components of routine renal care. Cardiovascular risk factors such as hypertension and dyslipidemia should be managed as per standard guidelines.Patients may require anti-anginal or antiarrhythmic therapies and management of heart failure. Gastrointestinal symptoms are managed in a similar way to irritable bowel syndrome. Depression and anxiety are common and may be managed in conjunction with a psychiatry team. Pain and acral paresthesias can be disabling for patients; often pain specialists are involved as part of the multi-disciplinary teams to help manage chronic neuropathic, acute noci-ceptive or inflammatory pain.
Surveillance may depend on the organ systems involved. For instance, blood and urine tests including quantification of proteinuria are performed routinely at each clinic review (this could vary from every 3 to 12 months depending on disease severity). Patients with cardiac involvement may require annual electrocardiograms, echocardiograms, Holter monitors and biennial cardiac MRIs. Patients with neurological involvement may require annual or biennial brain MRIs. Other annual evaluation of pain and QoL, audiometry and ophthalmology tests as indicated and routine blood and urine tests performed every 3-6 months.
Prognosis
The long term prognosis of patients with untreated Fabry disease is poor when compared to the general population. The median cumulative survival in untreated males and females is ~50 and ~70 years respectively. Cardiovascular disease is the most common cause of death followed by cerebrovascular and renal disease. The impact of Fabry specific therapy on survival remains unclear.
Summary
Fabry disease is a rare X-linked lysosomal storage disorder that involves reduced or deficient alpha-GAL enzyme activity, resulting in gradual accumulation of the glycosphingolipid GB3 and its derivatives in various organs with ensuing diverse clinical presentations. Severe classic forms occur in males. A detailed medical history and physical examination as well as multi-systemic diagnostic evaluation are parts of the initial work-up. Multi-disciplinary teams usually manage the multiple issues associated with the disease. Treatment often includes Fabry-specific therapy (ERT or chaperone therapy) and focuses on prevention of complications and improvement of QoL.
Excellent