MINI-REVIEW
IgA Vasculitis Associated with Nephritis: Mini-Review

By Dr Payal Gaggar, MD, DM
Assistant Professor
Department of Nephrology
Nizam’s Institute Of Medical Sciences
Hyderabad, India
GlomCon editors who contributed significantly to the development of this article include Paolo Nikolai So, MD, and Nasim Wiegley, MD.
Abstract
Kidney involvement occurs in 20%-80% of cases of IgA vasculitis (IgAV) and is responsible for the majority of observed morbidities. It can present as microscopic urinary abnormalities, nephritic syndrome, nephrotic syndrome, or acute renal failure. Its management depends on the severity of nephritis, with immunosuppressive therapy reserved for those developing rapidly progressive kidney failure. Its overall prognosis is favorable in children, whereas kidney involvement in adults may be severe with poor outcomes.
Introduction
First described by a London based physician named William Heberden in 1801, Henoch-Schönlein purpura (HSP) was further characterized by Johann Schönlein (1837) and Eduard Henoch (1874) to include arthritis, non-thrombocytopenic purpura, gastrointestinal manifestations and kidney involvement, hence the famous moniker. With a better understanding of the pathophysiology of the disease over the years, the International Chapel Hill Consensus Conference Nomenclature of Vasculitides in 2012 renamed HSP as immunoglobulin A vasculitis (IgAV). IgAV is an autoimmune vasculitis characterized by the formation of IgA-dominant immune deposits in the blood vessel walls (Table 1).
Table 1. Classification criteria for IgAV
| ACR 1990 | EULAR/PRES/PINTO |
|---|---|
2 or more criteria are required:
|
Mandatory criteria: palpable purpura without thrombocytopenia with lower limb predominance plus one of the four below:
|
IgAV is the most common form of vasculitis in children, with an incidence of 10-30/100,000 cases per year, usually running a self-limiting course. The peak age is six years (2-18 years). Kidney involvement is seen in a median of 30% of cases (20%-50%), with 91% occurring within the first six weeks and around 97% within the first six months. A lack of kidney involvement in the first six months predicts a low likelihood of progressive kidney diseases. Hence, urinalysis and blood pressure monitoring are necessary for at least six months, and optimally for 12 months from the initial presentation of systemic disease. The follow-up strategy may include weekly analysis for the first month, every other week for the second month and monthly for six months, then every three months for another six months. Women with a history of IgAV during childhood are at a high risk of complications such as proteinuria and hypertension during pregnancy, seen in around 70% of cases, mandating close monitoring during the antenatal period. The prognosis is favorable in 95% of cases, with ̴1-2% of children progressing to end-stage kidney disease (ESKD).
IgAV is relatively rare in adults, with an incidence of 0.8-2.2/10000 person-years. The median age of disease manifestation is 50 years, and in patients older than 60 years of age who have IgAV, evaluation for lung, kidney and prostate cancers should be considered. The development of kidney damage in adults is frequent and is seen in 45%-85% of cases with more severe disease than in children. There is a 13% risk of severe kidney failure and an 11% risk of progression to ESKD.
Pathogenesis
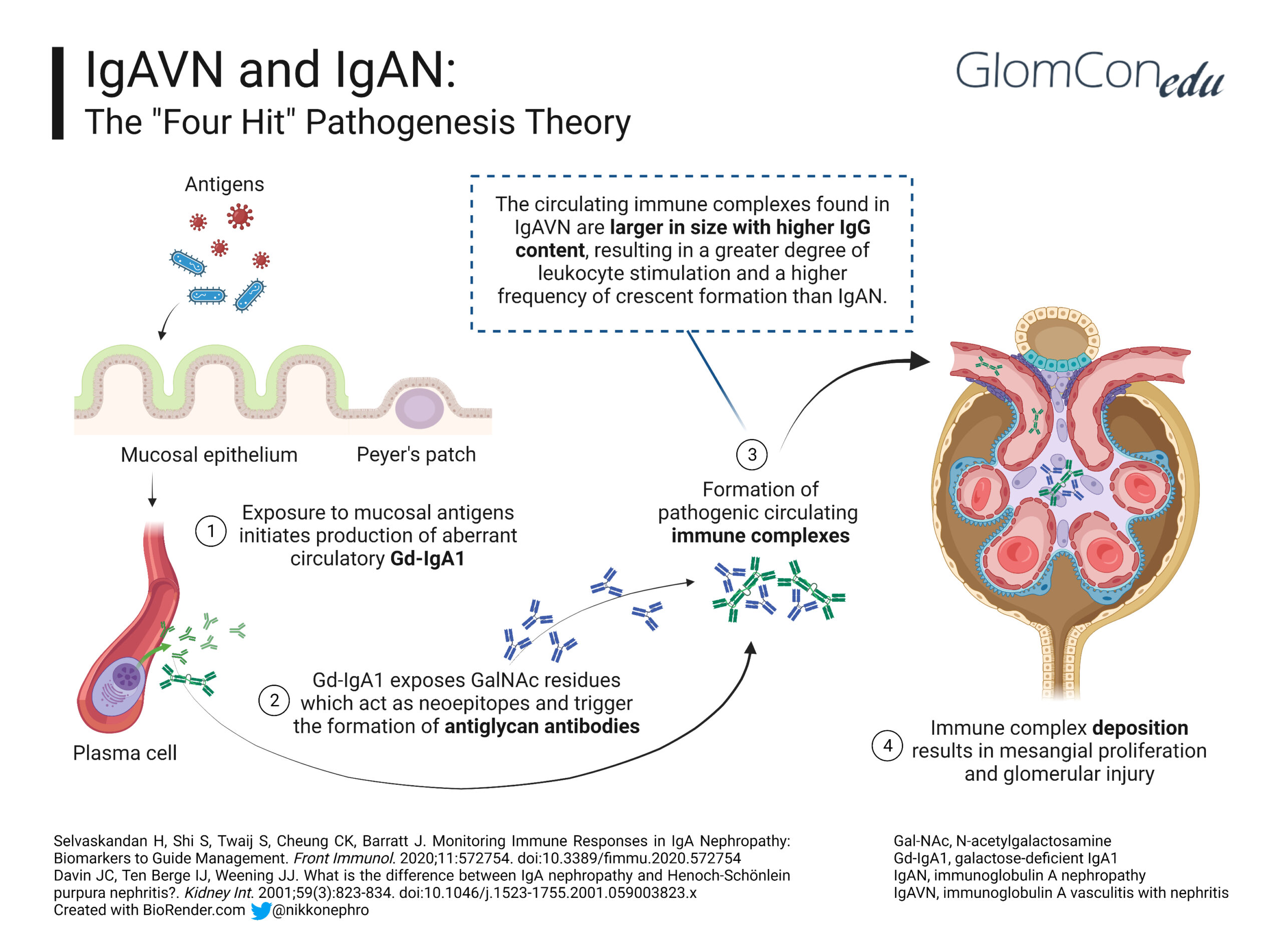
A history of upper respiratory tract infection or exposure to allergens in food, drugs (e.g., beta-lactamases, fluoroquinolones, macrolides, TNF-alpha blockers), insect bites and some vaccines (e.g., influenza, MMR, typhoid, cholera, and yellow fever) has been associated with the onset of IgAV. This finding suggests that infection or exposure to mucosal antigens may increase the production of the mucosal antibody galactose-deficient IgA1 (Gd-IgA1), thereby triggering the development of IgAV. Kidney involvement in IgAV, otherwise known as IgAV with nephritis (IgAVN), is postulated to follow the same four-hit hypothesis of IgA nephropathy (IgAN) (Figure 1):
Hit 1: Alterations in the O-linked glycosylation of IgA1 lead to the formation of Gd-IgA1.
Hit 2: The abnormal glycosylation exposes nearby GalNac residues, which act as neoepitopes and lead to IgG antibodies against Gd-IgA1.
Hit 3: Transferrin receptors (Tfr-CD71) are a group of IgA1 receptors expressed in mesangial cells. In IgAV nephritis, Tfr expression increases, leading to the deposition of Gd-IgA1–IgG circulating antibody complexes in the mesangium.
Hit 4: The immune complexes trigger mesangial proliferation, increased expression of proinflammatory cytokines and complement activation, augmenting the injury in the kidney.
Figure 1. The “Four-Hit” Pathogenesis Theory of IgAVN and IgAN.
Despite having similar mechanisms, the differences in the clinical presentation between IgAVN and IgAN (Table 2) are potentially due to increased IgE plasma levels and the larger size of circulating Gd-IgA1-IgG complexes with higher IgG content in IgAVN, resulting in a greater degree of leukocyte stimulation and more frequent endocapillary and extracapillary inflammation in such cases.
Table 2. Comparative features between IgAN, IgAVN and IgA-dominant IRGN
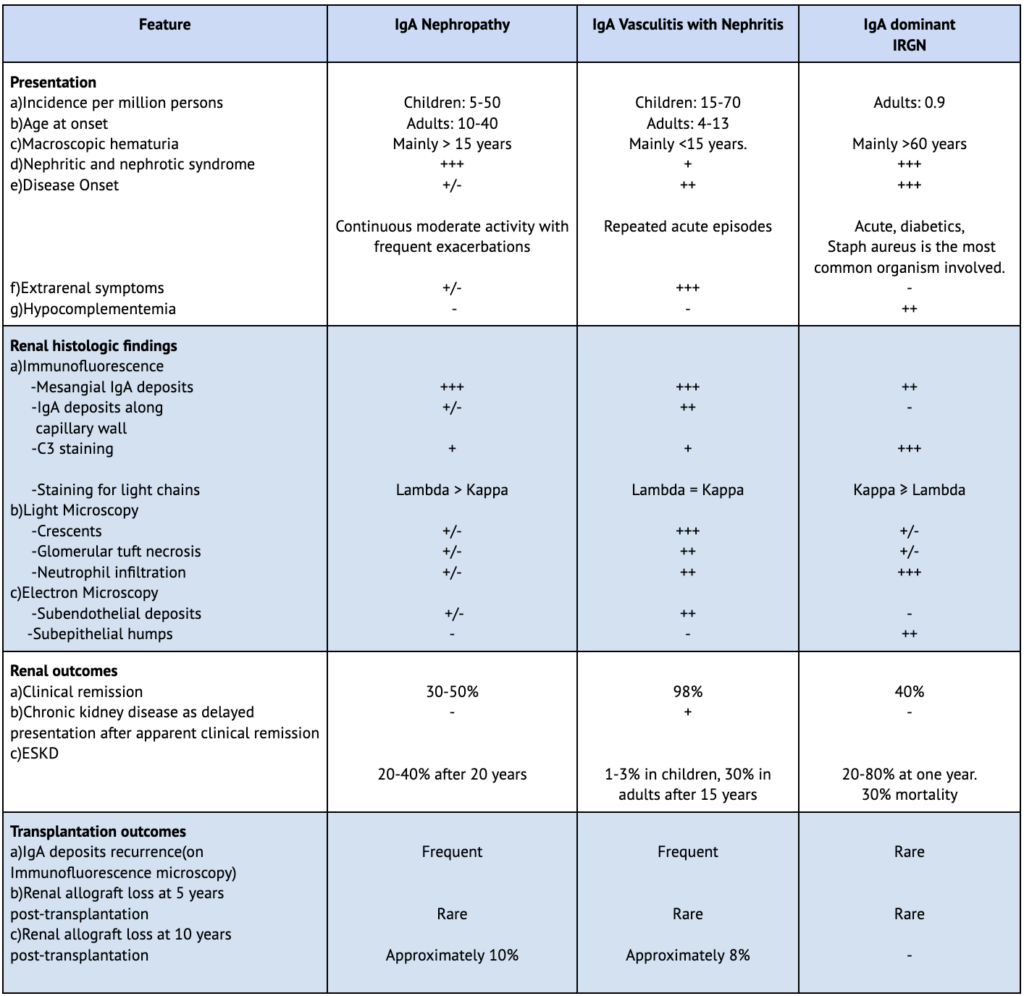
– , +/-, +, ++, +++ indicates the relative likelihood of a feature occurring directly comparing IgAN with IgAVN.
ESKD, end-stage kidney disease; IgAVN, IgA vasculitis with nephritis; IRGN, infection-related glomerulonephritis.
Clinical presentation
IgAVN has varied manifestations, from isolated microscopic hematuria or proteinuria to acute kidney injury. In a single series from a tertiary center studying long-term outcomes (mean of 23.4 years after onset) of individuals with a history of childhood IgAVN, microscopic or macroscopic hematuria with or without proteinuria were seen in 50% of patients, an acute nephritic syndrome in 8%, nephrotic syndrome in 13% and an association of nephritic and nephrotic syndromes in 29%.
Indications for renal biopsy in adults with a vasculitic rash typical of IgAV are as follows:
- Persistent and/or significant nephritis
- Rapidly progressive glomerulonephritis (RPGN), defined as ≥50% decline in eGFR over ≤3 months
- Proteinuria > 1g/day
- Impaired kidney function
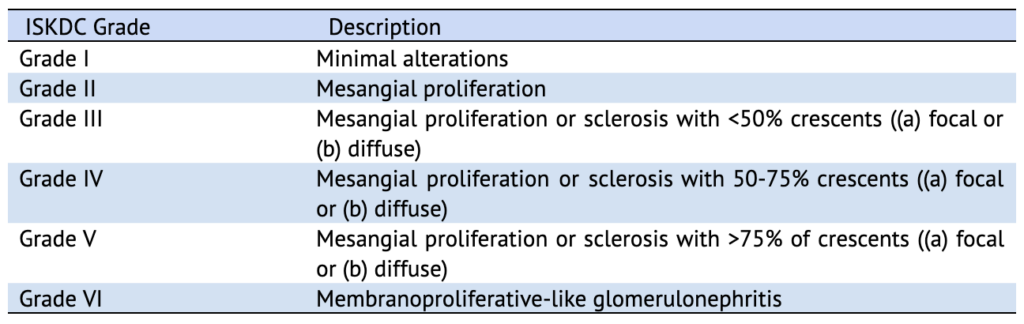
The International Study of Kidney Disease in Children (ISKDC) classification (Table 3) is the most frequently used grading system for IgAVN, which is based on the percentage of crescents in the kidney biopsy sample. But the presence or absence of crescents on kidney biopsy was not found to correlate with outcomes. The ISKDC classification majorly reflects ongoing active inflammation with no inclusion of tubulointerstitial or vascular compartments. The Oxford MEST-C scoring has not been validated in IgAVN.
Prevention of nephritis in IgAV
The Kidney Disease Improving Global Outcomes (KDIGO) guidelines do not recommend using glucocorticoids to prevent nephritis in children with IgAV without kidney involvement. A meta-analysis of 5 RCTs in which 789 children with IgAV were examined for the effects of short-duration glucocorticoids (2-4 weeks) on preventing persistent nephritis at 6 and 12 months after the presentation concluded that such treatment with glucocorticoids at presentation had no preventive effect on the onset of persistent nephritis. There are no such similar RCTs in adults.
IgAVN
Patients with proteinuria >0.5g/day should be treated with maximally tolerated doses of ACEi/ARBs. SGLT2 inhibitors, despite being associated with a reduction in proteinuria in patients with IgAN, still require further studies to explore their potential role in IgAVN. The International IgAN Prediction tool is also not designed for prognostication in IgAVN. Of note, the presence of crescents on kidney biopsy in patients without any clinical signs of RPGN is not an automatic indication for the commencement of immunosuppression. No specific dietary intervention has been shown to alter outcomes in IgAVN. Lifestyle modifications like weight control and smoking cessation should be advised.
In contrast, patients with proteinuria of >1g/day, despite treatment with a maximally tolerated dose of the renin-angiotensin system (RAS) blockade for three months, are considered to be at high risk of progression to chronic kidney disease. As such, they may be considered for adding glucocorticoids as used for IgAN after a detailed discussion of the risks and benefits of immunosuppression with the patient.
IgAVN with RPGN
IgAVN patients with RPGN should be treated in accordance with the KDIGO guidelines for ANCA-associated vasculitis (AAV). Induction may consist of a regimen containing methylprednisolone pulses followed by oral corticosteroids and intravenous cyclophosphamide, whereas maintenance therapy of low-dose oral steroids combined with azathioprine or mycophenolate mofetil. The duration of induction and maintenance phases of treatment, recommendations for weaning treatment, and methods in monitoring therapeutic response remain uncertain due to the lack of large randomized controlled trials RCTs in IgAVN.
Literature on the use of other immunosuppressive agents is uniquely heterogeneous as they involve different disease definitions and classification criteria. Rituximab has shown benefit in a few case series on children with severe and relapsing skin and kidney involvement refractory to other therapy. An uncontrolled case series describes the potential role of the addition of plasma exchange to glucocorticoid therapy in accelerating the recovery of patients with life- and organ-threatening complications of IgAV, including severe kidney involvement. Early initiation of plasma exchange monotherapy in children with IgAVN with RPGN was observed in a case series to effectively improve their long-term prognosis regarding the course of proteinuria and progression to end-stage kidney failure.
In contrast to the KDIGO guidelines, the SHARE (Single Hub and Access point for pediatric Rheumatology in Europe) initiative (Table 4) recommends the use of immunosuppression in all cases of IgAVN irrespective of the RPGN presentation. This further highlights the heterogeneous nature of IgAVN management worldwide, pending large evidence-based RCTs in the field.
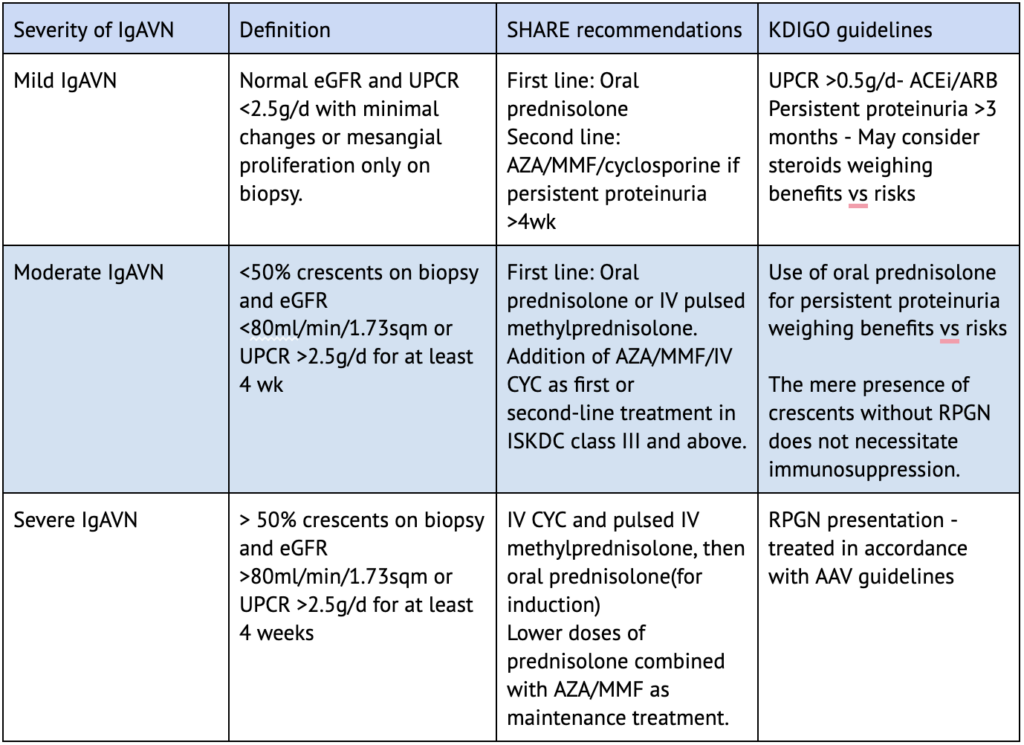
ACEi, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; AZA, azathioprine; CYC, cyclophosphamide; MMF, mycophenolate mofetil; UPCR, urine protein-creatinine ratio.
Prognosis and follow-up
The risk of long-term kidney impairment in children is low (1.6%) particularly in those with isolated proteinuria or hematuria but is much higher (19.5%) if the initial presentation is nephritic or nephrotic syndrome. ESKD develops in 10 to 30% of adults at 15 years from the time of diagnosis. Factors associated with evolution to ESKD include baseline renal function, baseline proteinuria >1g/day, macroscopic hematuria, hypertension, and proteinuria >1g/day during follow-up. Kidney biopsy findings of interstitial fibrosis, sclerotic glomeruli, and fibrinoid necrosis are also associated with poor prognosis.
Conclusion
IgAV is generally a self-limiting disease, with kidney involvement occurring in 20%-50% of children and in 45%-85% of adults. In children the disease is usually self-limiting. Among adults , those with higher baseline proteinuria, hypertension, or biopsy features of chronicity have a poorer prognosis of kidney disease. IgAVN is generally managed with supportive care, with immunosuppression reserved for severe cases (e.g., RPGN). IgAVN represents an area of unmet research needs, requiring randomized controlled trials to determine the most appropriate course of action and improve patient outcomes.