DIAGNOSTIC WORKUP
Genomics in Glomerular Disease: From Bench to Bedside

By Dr Benjamin Tan, MB BCh BaO (NUI), MRCP (UK)
Nephrologist
Queen Elizabeth Hospital, Kota Kinabalu, Sabah
GlomCon editors involved in the development of this article: Paolo Nikolai So MD, Nasim Wiegley MD
Introduction
Genetic kidney disease (GKD) is a known cause of end-stage kidney disease (ESKD), accounting for up to 10% of cases in adults and nearly 70% in children with chronic kidney disease (CKD). Causes of GKD include cystic-related kidney disorders, glomerular diseases, congenital abnormalities of the kidney and urinary tract (CAKUT) and metabolic nephropathies. The European Rare Kidney Disease Registry (ERKReg) and the National Registry of Rare Kidney Diseases (RaDaR), under the auspices of The UK Kidney Association (UKKA), are just some of the few regional registries that have been established to collect epidemiological data on GKD.
Genetic glomerular disease can be broadly thought of to be either monogenic or polygenic. Monogenic disorders refer to diseases caused by rare, pathogenic variants in a single gene. In contrast, polygenic disorders refer to diseases caused by a cumulative effect of common genetic variants superimposed with environmental factors. The actual global burden of monogenic glomerular disease is currently underestimated. A recent study showed that almost 35% of CKD patients diagnosed with GKD based on a positive genetic test result had a diagnosis of monogenic glomerular disease. This review summarizes the current understanding of the pathogenetic and application of clinical genomics to diagnose monogenic glomerular disease.
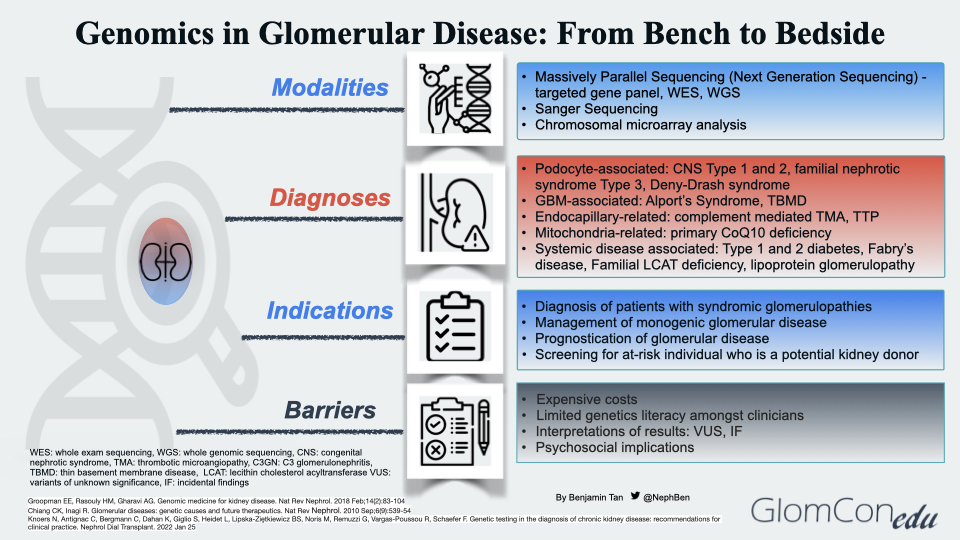
Modalities of Genetic Testing
Massively parallel sequencing (MPS), known as next-generation sequencing (NGS), is the main technique used in the genomic sequencing of glomerular diseases. This technique involves parallel sequencing of multiple small fragments of DNA to determine the sequence. Targeted phenotype-associated gene panels using NGS technology are performed as the precise etiology of monogenic glomerular disease may remain unclear despite extensive history taking, blood investigations, imaging, and histopathological studies. They allow rapid identification and diagnosis in a cost-effective manner. If the targeted gene panel result is negative, other panels with broader coverage content may be used. More comprehensive tests, such as whole exome sequencing (WES) or whole genome sequencing (WGS), are then carried out. Table 1 summarizes the different genetic testing modalities with their respective indications, advantages and disadvantages.
Table 1. Genetic testing modalities, indications, advantages and disadvantages
| Modality | Scope of Analysis | Indications for Use | Advantages | Disadvantages |
|---|---|---|---|---|
| Targeted gene panel (NGS) | Identification of SNVs and small INDELs (<1kb) within gene of interest for clinical phenotype | Patients with phenotypes related to a specific diagnosis and with low genetic heterogeneity | Analysis of all genes related to a particular phenotype
Emphasis on genotype-phenotype relationship and minimize risk of secondary findings |
Limited capacity for sequence reanalysis
Challenges with panel design (need updates) Short reading length (50-300bps) |
| Whole exome sequencing (WES) | Identification of SNVs and small INDELs (<1kb) within coding region of genome | Patients left undiagnosed by NGS
Patients with genetically heterogenous/ non-specific phenotype |
Analysis of all coding regions in genome
Unbiased approach increases diagnostic sensitivity Allows for sequence reanalysis and discovery of new genes |
Challenges with interpretation of secondary findings
Multiple candidate variants challenging and time consuming Less accurate for INDELs Limited coverage of repetitive regions |
| Whole genome sequencing (WGS) | Identification of SNVs and small INDELs (<1kb) within coding and non-coding regions of genome | Patients undiagnosed by all other modalities
Patients with highly genetically heterogeneous phenotypes Patients with non-specific phenotypes |
Superior to WES in terms of ability to assess SNVs, INDELs, CNVs in coding and non-coding regions
Useful for sequence reanalysis Can detect deep splicing and intronic variants not picked up by other techniques |
Difficulty to interpret non-coding variants
Delay in return of results due to prolonged interpretation of overwhelming genetic data Detection of secondary findings Burden of storage of long term sequence data |
| Sanger Sequencing | Detection of small variants (SNVs/INDELs <10bp) within a DNA segment of <1kb | Confirmation of NGS identified variants
Confirmation of a mutation in a specific gene Where NGS is inadequate for a diagnosis ie. highly repetitive segments, high GC content |
High analytical accuracy
Fast and easy interpretation resulting in high turnaround time No risk of secondary findings |
Unable to detect large structural variants ie CNVs
Time- and cost-inefficient for analysis of large segments of DNA |
| Chromosomal microarray | Detection of small chromosomal rearrangements/CNVs (>200-400kb) | Phenotypes with large genomic rearrangements ie multiple congenital anomalies | Higher resolution allows identification of CNVs missed by standard karyotyping | Cannot detect SNVs, INDELs and small CNVs (<200kb)
Limitations in detecting balanced chromosomal rearrangements, CNVs in repetitive segments |
Abbreviations: CNV, copy nucleotide variant; DNA, deoxyribonucleic acid; GC, glucocorticoid; INDEL, insertion-deletion; SNV, single nucleotide variant.
Genetic Diagnoses of Glomerular Disease
The genetic diagnoses of glomerular diseases can be classified based on the anatomical and pathological involvement within the glomerulus (Figure 2). Podocyte-associated genetic defects affect the slit diaphragm function and disrupt the actin-cytoskeleton framework within the podocyte foot processes that anchor to the glomerular basement membrane (GBM). Genetic defects may also be seen in several GBM components, such as the cross-linkage network of type 4 collagen chains embedded within triple-helical protomers as well as laminin. In endocapillary-related genetic defects, a prothrombotic state ensues, leading to thrombotic microangiopathy (TMA), a histopathological feature commonly observed in both thrombotic thrombocytopenic purpura and atypical HUS (complement-mediated TMA). Mutations in mitochondrial genes may also result in glomerular disorders. Using a Drosophila model to investigate the relationship between COQ2 mutation and structural changes within the podocyte, Zhu et al. suggested the role of oxidative stress injury in podocytes as a postulated mechanism of glomerular injury in patients with coenzyme Q10 (CoQ10) deficient-related glomerular disorders, otherwise known as primary CoQ10 nephropathy. To date, these mutations (PDSS1, PDSS2, COQ2, COQ6 and COQ8B/ADCK4) within the CoQ10 biosynthesis pathway have been associated with glomerular involvement. Lastly, the effects of systemic genetic diseases on the glomerular filtration barrier are thought to be indirect, ranging from disruption of normal cellular metabolism within the glomerular cells to excessive deposition of substances such as lipoproteins and immune complexes within the glomerulus.
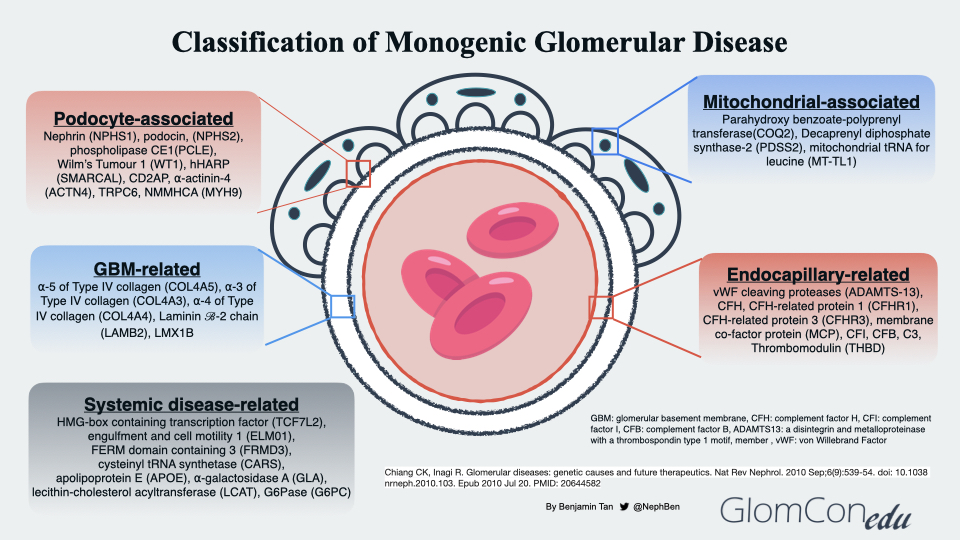
Figure 2. Classification of monogenic glomerular dIsease based on the anatomical and pathological involvement within the glomerulus.
Potential Indications for Genetic Testing
a) Diagnosis of patients exhibiting features of syndromic glomerulopathies
The diagnostic performance of genetic testing in monogenic glomerular disease correlates inversely with age of onset, ranging from almost 100% at birth to about 50-70% in children and young adults. These disorders present with a variety of extra-renal clinical features and are resistant to standard glucocorticoid therapy (Table 2).
Table 2. Syndromic glomerulopathies based on the age of onset and predominant extra-renal features
| Age of Onset | Predominant extra-renal features | Conditions |
|---|---|---|
| Neonate | Neurological | Galloway-Mowat syndrome, Leigh Syndrome with nephrotic syndrome |
| Cutaneous | Hypotrichosis-lymphedema-telangiectasia-renal defect syndrome, nail-patella syndrome | |
| Ocular | Pierson syndrome | |
| Auditory | MYH-9 related disease, familial SRNS with sensorineural hearing loss | |
| Genitourinary | Denys-Drash syndrome | |
| Endocrine | Familial SRNS with adrenal insufficiency, fibronectin glomerulopathy | |
| Musculoskeletal | Schimke immuno-osseous dysplasia | |
| Respiratory | Congenital nephrotic syndrome-interstitial lung disease-epidermolysis bulla syndrome | |
| Infant | Neurological | Galloway-Mowat syndrome, severe oculorenal cerebellar syndrome, Leigh Syndrome with nephrotic syndrome |
| Cutaneous | Hypotrichosis-lymphedema-telangiectasia-renal defect syndrome, nail-patella syndrome | |
| Ocular | Pierson syndrome | |
| Auditory | MYH-9 related disease, familial SRNS with sensorineural hearing loss | |
| Genitourinary | Denys-Drash syndrome | |
| Endocrine | Familial SRNS with adrenal insufficiency, fibronectin glomerulopathy | |
| Musculoskeletal | Schimke immuno-osseous dysplasia | |
| Childhood | Neurological | Galloway-Mowat syndrome, autosomal dominant intermediate, Charcot-Marie-Tooth disease Type E |
| Cutaneous | Nail-patella syndrome | |
| Ocular | Pierson syndrome | |
| Auditory | Alport syndrome, MYH-9 related disease | |
| Genitourinary | Denys-Drash syndrome, Frasier syndrome | |
| Endocrine | Familial SRNS with adrenal insufficiency, fibronectin glomerulopathy, lipoprotein glomerulopathy | |
| Adolescent | Neurological | Action myoclonus-renal failure syndrome, autosomal dominant intermediate, Charcot-Marie-Tooth disease Type E |
| Cutaneous | Nephrotic syndrome-epidermolysis bullosa-sensorineural hearing loss syndrome | |
| Auditory | Alport syndrome, MYH-9 related disease | |
| Genitourinary | Denys-Drash syndrome, Frasier syndrome | |
| Musculoskeletal | Schimke immuno-osseous dysplasia | |
| Endocrine | Fibronectin glomerulopathy, lipoprotein glomerulopathy | |
| Young Adulthood | Neurological | Action myoclonus-renal failure syndrome, autosomal dominant intermediate, Charcot-Marie-Tooth disease Type E |
| Auditory | Alport syndrome, MYH-9 related disease | |
| Genitourinary | Frasier syndrome | |
| Endocrine | Fibronectin glomerulopathy, lipoprotein glomerulopathy | |
| Musculoskeletal | Schimke immuno-osseous dysplasia |
Abbreviations: MYH-9, myosin heavy chain 9; SRNS, steroid-resistant nephrotic syndrome.
b) Management of monogenic glomerular disease
Monogenic variants in over 60 genes have been identified as the cause of steroid-resistant nephrotic syndrome (SRNS; defined as lack of complete remission at 4 weeks of therapy with daily prednisolone at standard dose) in approximately 30% of pediatric patients. As such, genetic testing should be considered in children, adolescents and young adults who develop SRNS, prioritizing those with a strong family history of proteinuria, hematuria or CKD of unknown origin. It may then help clinicians decide whether to continue immunosuppression in patients with steroid-resistant or partially responsive nephrotic syndrome. Such testing can provide a convenient and cost-effective method of diagnosis while minimizing the risks involved in kidney biopsy, and avoiding the pitfalls of misdiagnoses, potentially harmful diagnostic procedures and incorrect treatments.
A confirmed genetic diagnosis also leads to the delivery of a more targeted therapy, such as enzyme and CoQ10 replacement for Fabry’s disease and CoQ10 deficient SRNS, respectively. In Alport syndrome (AS), it enables early initiation of renin-angiotensin-aldosterone system blockade which is beneficial in slowing down disease progression in the kidneys. In patients with complement-mediated TMA (CM-TMA), a negative genotyping result is associated with a low risk of relapse, potentially allowing the discontinuation of C5 inhibitor therapy such as eculizumab.
c) Prognostication of glomerular disease
Genetic diagnosis of AS helps prognosticate disease course and progression. X-linked (COL4A5) and autosomal recessive forms of AS (COL4A3/4) typically present with progressive kidney dysfunction and bilateral sensorineural hearing loss, while autosomal dominant forms of AS (COL4A3/4) present with a milder form of progression without hearing loss.
Complement gene testing provides information regarding the risk of disease recurrence in kidney allografts post-transplantation. In the pre-eculizumab era, a confirmed pathogenic variant in CFH, CFB, TBHD or CFH:CFHR1 rearrangements conferred a high recurrence risk of almost 80-90% while a moderate recurrence risk between 40-75% was observed in those with a negative complement genotyping study or confirmed isolated CFI or C3 mutation. An isolated MCP or DGKE mutation conferred a low recurrence risk of less than 20%. According to a systematic review and meta-analysis on the outcomes of patients with CM-TMA who were treated with prophylactic eculizumab post-kidney transplantation, less than 6% of patients experienced disease recurrence when eculizumab therapy was stopped.
d) Screening for an at-risk individual who is a potential kidney donor
In the setting of living-related kidney transplantation, genetic testing offers a non-invasive way of screening potential donors at risk of developing a hereditary glomerular disease. Individuals with a confirmed pathogenic variant are usually excluded from kidney donation. Genetic testing of at-risk family members enables early disease detection and allows future family planning by clarifying inheritance patterns.
Barriers
Genetic testing faces multiple barriers to access and use. Whereas genetic testing is inaccessible in most developing countries, such services have high costs and long turnaround times, often with more accessible coverage for children than adults in countries offering reimbursements. There is limited data on the cost-effectiveness of genetic testing in managing glomerular diseases, with a few prospective studies demonstrating cost-effectiveness amongst children with suspected monogenic kidney disorders.
Limited genetics literacy amongst nephrologists is another major barrier. Genetics is seldom incorporated into the curriculum of nephrology fellowship training programs in many countries, making the analysis and interpretation of test results challenging for the nephrologist. A recent survey conducted to explore the attitude of nephrologists toward genomics showed that even though the majority believed in the usefulness of genetic testing, only less than a quarter felt confident to use the results. Interpreting the sequence data requires gene- and variant-level analyses, phenotypic-level comparison and determination of actionability. Commonly encountered terminologies such as variants of undetermined significance and incidental findings can be challenging to interpret and may hence require the expertise of a clinical geneticist or a trained genetics counselor.
Another challenging aspect is the psychosocial implications associated with genetic testing. Stress and anxiety may arise among patients, followed by stigmatization from their families or community members who may not comprehend their genetic diagnosis. Patient insurability and confidentiality may also be affected despite laws being legislated to protect patients from being discriminated against by employers and insurance companies.
Conclusion
The future of nephrogenetics in glomerular disease is bright. Recent advances in genomics technology, data collection and experimental research models have paved the way for a greater understanding of the pathogenetic and molecular mechanisms of monogenic glomerular disease. Such understanding may allow the development of specific therapeutic targets that are safe and effective. With precision medicine on the horizon, we must be cognizant of the potential challenges in bridging translational research to clinical medicine.
Visual Abstract