TREATMENT
Rituximab Resistance in Glomerular Diseases

By Dr. Tania Salehi
Nephrology & Clinical Immunology Advanced Trainee
Royal Adelaide Hospital
Adelaide, South Australia
Edited by Anoushka Krishnan, Ayman Al Jurdi, and Nasim Wiegley.
B-cell depletion therapy with rituximab has been increasingly utilized in the management of various immune-mediated glomerular diseases, both newly diagnosed and relapsing diseases. This agent is a chimeric mouse-human monoclonal antibody (mAb) made up of a murine variable region directed against the CD20 antigen on B lymphocytes. It induces B-cell depletion through various mechanisms, including Fc receptor 𝛾-mediated antibody-dependent cytotoxicity (ADCC), complement-mediated cell lysis, B-cell growth arrest and B-cell apoptosis.
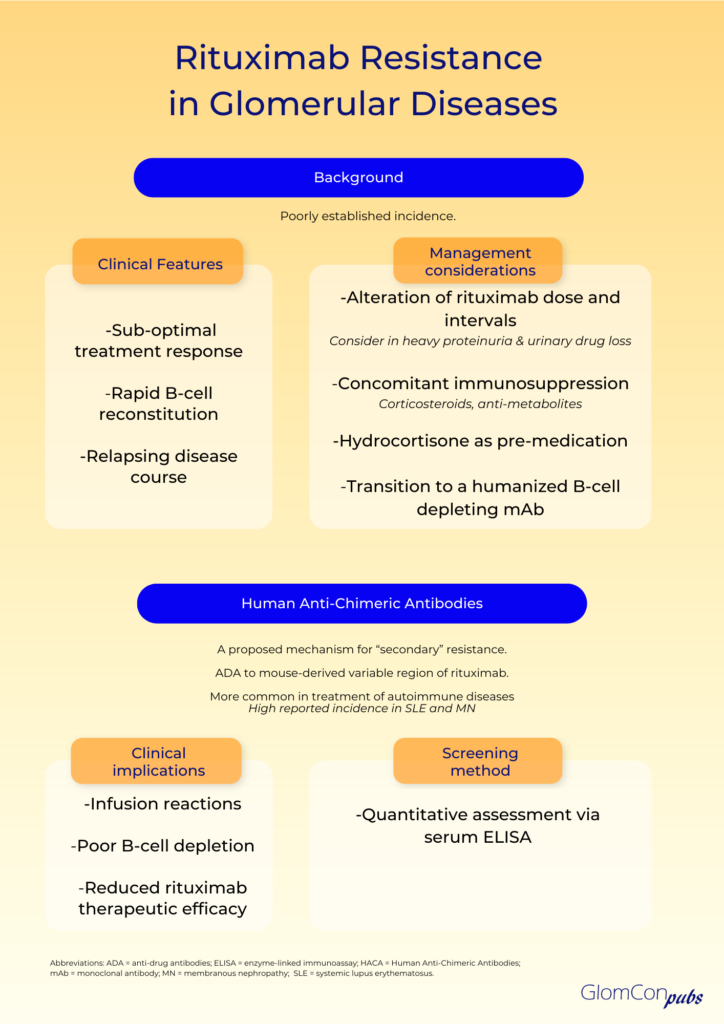
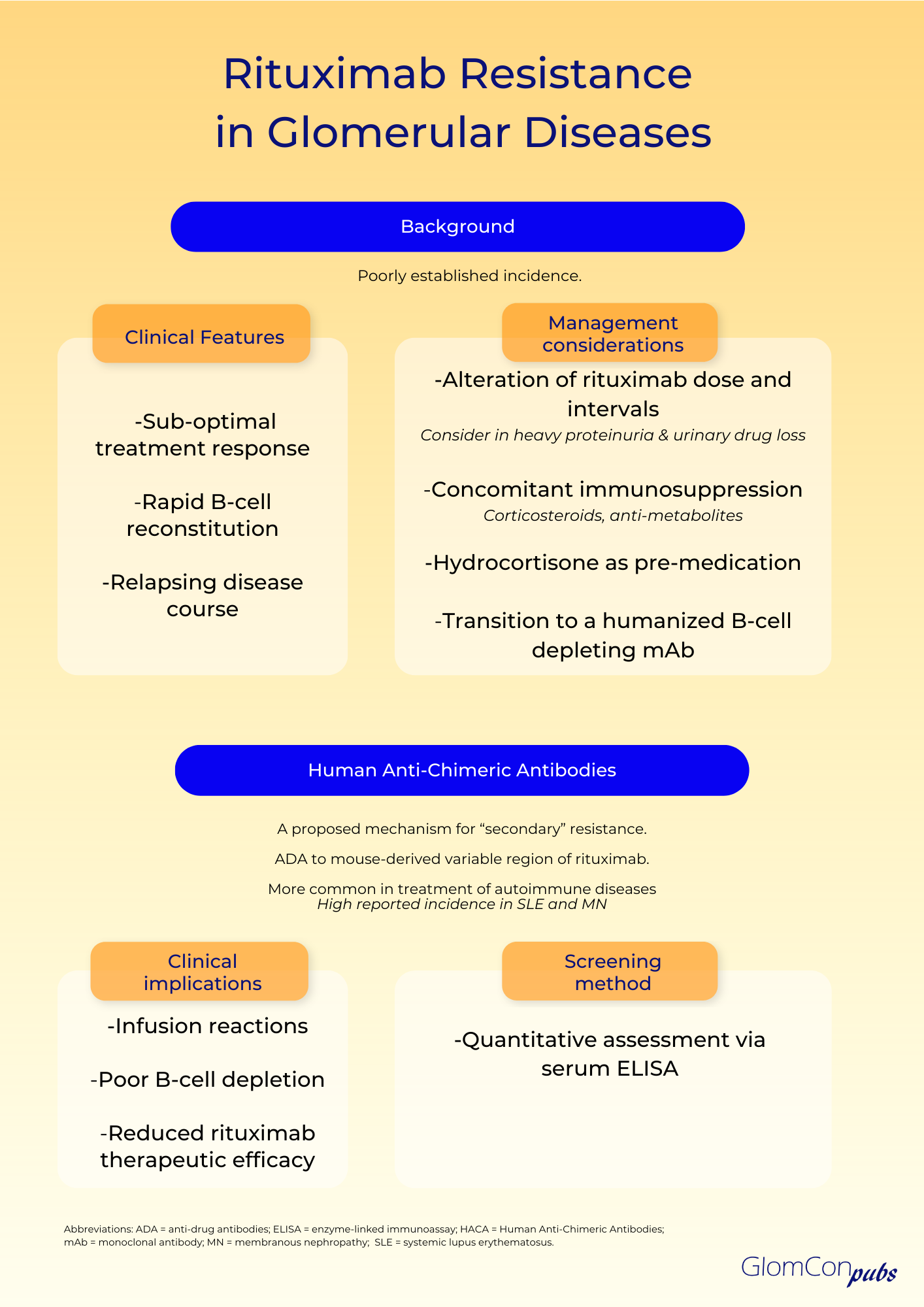
Resistance to rituximab is a clinically pertinent adverse sequela, which is defined as a lack of response to rituximab-containing regimens, in particular, disease progression within 6 months of therapy. The true incidence remains unknown. Clinical suspicion of rituximab resistance is important in individuals with relapsing disease, suboptimal treatment response and rapid B-cell reconstitution. It is more commonly observed in autoimmune diseases, such as systemic lupus erythematosus (SLE), membranous nephropathy and rheumatoid arthritis. The mechanism remains uncertain, and numerous hypotheses have been proposed.
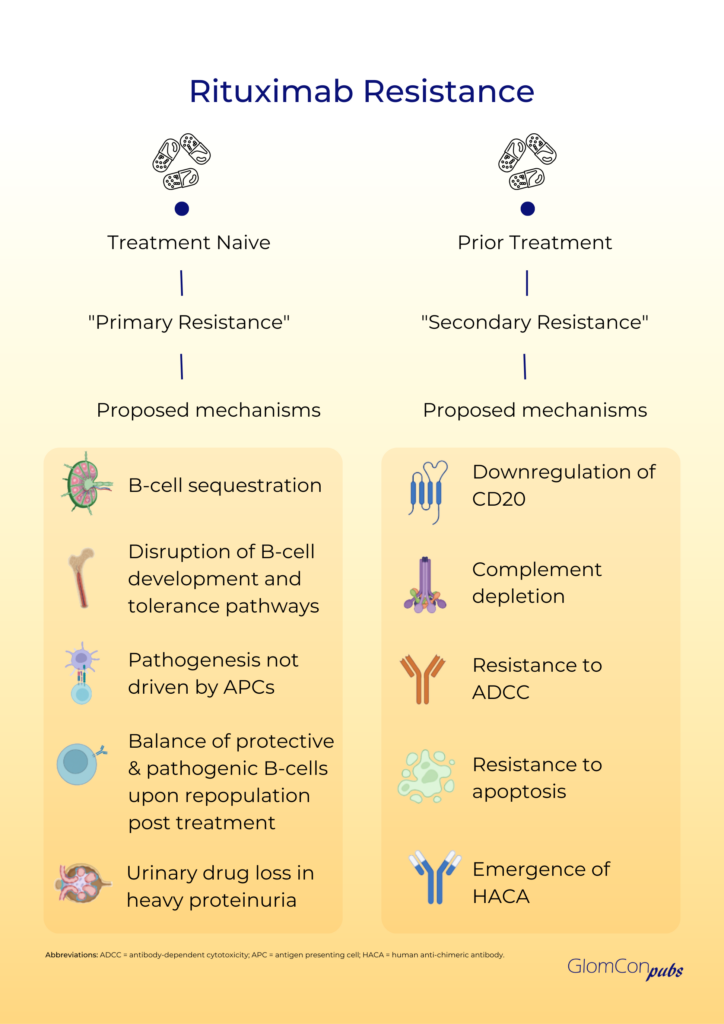
Primary rituximab resistance has been observed in treatment-naïve individuals. The cause is poorly understood; however, B-cell sequestration and disruption of B-cell development and tolerance pathways may be contributory. The balance of protective and pathogenic B-cell subsets established following B-cell repopulation are also significant. In addition, the underlying disease pathophysiology is relevant to primary resistance, particularly when antigen-presenting cells do not play a critical role in pathogenesis. Furthermore, rapid urinary loss of rituximab in heavy proteinuric states is likely to yield a suboptimal treatment response. This has been illustrated in the LUNAR trial, where the addition of rituximab to lupus nephritis induction therapy did not influence remission rates. Secondary analysis of the LUNAR trial displayed that heavier proteinuria at the time of rituximab treatment was associated with lower peak rituximab levels, incomplete peripheral B-cell depletion and lower odds of clinical response to rituximab.
Secondary rituximab resistance occurs with repeated drug exposure. Here, the major effector pathways of rituximab’s mechanism of action are involved. Such pathways include the downregulation of CD20, complement depletion, resistance to ADCC and resistance to apoptosis. Additionally, anti-drug antibodies (ADAs) may form, which not only target the mAb binding domain but can also alter drug pharmacokinetics via the formation of immune complexes and increased mAb metabolism. The emergence of human anti-chimeric antibodies (HACA) to the mouse-derived variable region of rituximab is a recognized consequence of rituximab therapy. These ADAs may contribute to increased infusion reactions, poor B-cell depletion and reduced therapeutic efficacy of rituximab by enhanced drug clearance. Enzyme-linked immunoassays (ELISA) can be used for the quantitative assessment of serum rituximab and anti-rituximab antibodies. Serial testing may be performed to monitor serum levels.
The incidence of HACA to rituximab remains uncertain, however, they are more commonly observed in the management of autoimmune diseases compared to hematological malignancies. This could be associated with a highly activated B-lymphocyte status and a tendency of developing ADA in the setting of underlying autoimmunity. The emergence of ADAs is notably high in SLE, where polyclonal B cell activation is present. An incidence of 37.8% has been reported, most commonly in the setting of lupus nephritis, a more active disease and a high SLEDAI-2 K score. Rituximab HACA has also been described in the treatment of membranous nephropathy, with reports as high as 23%. Here, the ADAs have been drug-neutralizing and associated with faster B-cell reconstitution and higher relapse rates. However, these findings have not been demonstrated in all glomerular diseases, as another study found no association between the development of ADAs and relapse in steroid-dependent nephrotic syndrome.
There is little evidence on how to circumvent rituximab resistance. Alterations of dose and reduction of treatment intervals may have a role, especially in those with rapid urinary drug loss. However, the optimal strategy remains to be defined. Concomitant therapy with other immunosuppressive agents, such as corticosteroids and anti-metabolites may theoretically diminish the development of HACA; an observation noted with infliximab mAb treatment. Pre-medication with intravenous hydrocortisone may also be of benefit. Most importantly however, transition to alternative B-cell depleting agents may be required. The rate of immunogenicity and cross-reactivity of rituximab HACA with other B-cell depleting agents is unknown. Type II anti-CD20 molecules, such as obinutuzumab, are directed against an alternative epitope and cause B-cell depletion primarily by apoptosis rather than complement-dependent cytotoxicity. Furthermore, the humanized nature of this mAb negates the risk of HACA development. As such, humanized anti-CD20 mAbs should be considered as alternative treatment options in those with rituximab resistance.
Conclusion
Increased awareness of the immunogenic consequences of rituximab administration is required among clinicians. Urinary losses of rituximab in the setting of heavy proteinuria may lead to suboptimal treatment response and rapid B-cell return, which may improve with more frequent rituximab infusions. Screening for HACA should be considered in those with high-risk autoimmune diseases, such as SLE, those demonstrating poor clinical response to therapy, more rapid B-cell reconstitution and relapsing disease. This is particularly significant if rituximab re-treatment is considered. Detection of HACA may assist in directing disease treatment options, with consideration of additional immunosuppressive therapy and alternative B-cell depleting agents.
Disclaimer: The author reports no conflicts of interest